(Image courtesy: Nishant Raj)
The effectiveness of any drug molecule depends on how well it interacts with the internal environment inside our body. Its pharmacokinetic (PK) properties determine how successfully it escapes degrading enzymes as it travels through the digestive system or the bloodstream, crosses biological barriers like the cell membrane, and reaches the desired target.
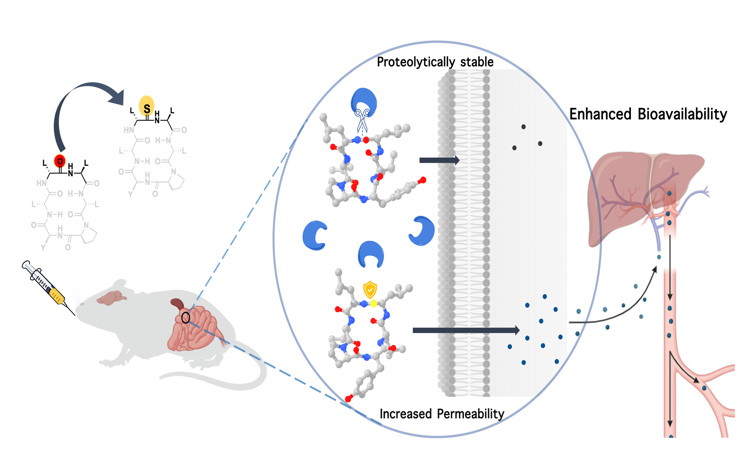
In a study published in Nature Communications, researchers at the Molecular Biophysics Unit (MBU) describe a novel method for improving the pharmacokinetic properties of “macrocyclic peptides” – drug molecules that are pursued heavily by pharmaceutical industries worldwide. The IISc team, in collaboration with Anthem Biosciences, has demonstrated that substituting just a single atom – oxygen with sulphur – in the backbone of a macrocyclic peptide can make it more resistant to digestive enzymes, and can increase its permeability through cell membranes, boosting its bioavailability.
A vast majority of today’s medicines are made up of small molecules taken orally in the form of pills. Larger molecules like monoclonal antibodies are much more specific and effective, but they must be injected. Scientists have, therefore, turned to macrocyclic peptides – chains of amino acid residues attached to each other via amide bonds, which are engineered to form circular structures. These compounds combine the best of both small and large pharmaceutical molecules.
However, like any protein, macrocyclic peptides are highly susceptible to digestive enzymes. They also find it hard to cross cell membranes which are made up of lipids, because they are water-loving molecules. The amide (CO-NH) bonds in these peptides interact with surrounding water molecules via relatively weaker bonds called hydrogen bonds. “For peptides to pass through a lipid membrane, they must reduce their hydrogen bonding with water. They must become a little more oil-loving (lipophilic),” explains Jayanta Chatterjee, Professor at MBU and corresponding author of the study. “Currently there are no concrete methods available apart from N-methylation to improve the pharmacokinetic properties of macrocyclic peptides,” he says.
Pritha Ghosh, former PhD student at MBU and first author, explains that the current N-methylation strategy requires exchanging a hydrogen atom from the amide bond with a methyl group. This prevents hydrogen bond formation between the nitrogen atom from the amide bond and the surrounding water, making it easier for the peptide to pass through the lipid membrane. However, such a modification has been shown to affect the binding of the peptide to its target, by making it too flexible and less specific. To overcome this drawback, Chatterjee and his team decided instead to focus on the oxygen atom in the amide bond, which is known to interact with two water molecules via hydrogen bonds. Using chemically synthesised cyclic peptides, they show that replacing this oxygen atom with sulphur makes the peptide much more lipophilic, increasing its permeability through the lipid membrane. They also found that this modification made the peptide less susceptible to digestive enzymes, since these enzymes are known to target the oxygen atom in the amide bond – which has now been swapped with sulphur.
To test whether such a modified compound can retain its biological function, the team used a shorter version of somatostatin – a hormone secreted by the pancreas that inhibits the growth hormone in our body – in which they substituted the oxygen atom of an amide/peptide bond with sulphur. The team found that when injected under the skin of model animals, the modified somatostatin not only lasted longer in the bloodstream than the unmodified one, but also effectively inhibited the growth hormone.
Ghosh says, “[After somatostatin], our lab continues to work with other biologically active molecules. Oxygen-to-sulphur modifications may be used in combination with other strategies … more than one substitution may give better results. We can use this technology to make peptides with better pharmacological properties.”





